 首页
首页DFT揭秘氧还原反应(ORR)机理|从吸附能到台阶图的深度解析
氧还原反应(Oxygen Reduction Reaction, ORR)是燃料电池和金属-空气电池的核心反应,但其多电子转移过程的复杂性使得催化剂设计极具挑战。本文将结合密度泛函理论(DFT)计算,深度解析ORR反应机理、活性描述符与催化剂设计原则,助你从原子尺度理解催化本质!
一、ORR反应路径与关键中间体
ORR在酸性/碱性环境中的反应路径相似,以酸性条件为例:
1.O₂吸附:O₂分子吸附于催化剂表面(*+ O2→O2*)。
2.质子与电子耦合:O2*与质子和电子耦合生成OOH*中间体。
3.脱附生成H₂O:OOH*进一步加氢生成O*和H₂O。
4.重复加氢再生成H2O:O* + 2H++2e-→* + H2O (实际计算过程HO*不可忽略)
关键中间体:
·O₂*:吸附态氧气分子
·OOH*:超氧中间体
·O*:原子氧吸附态
·OH*:羟基吸附态
吉布斯自由能(ΔG):各步骤的ΔG决定反应能垒,ORR是放热反应,理想催化剂需平衡各步骤能量(U=0V时,ΔG最优为-1.23eV)。
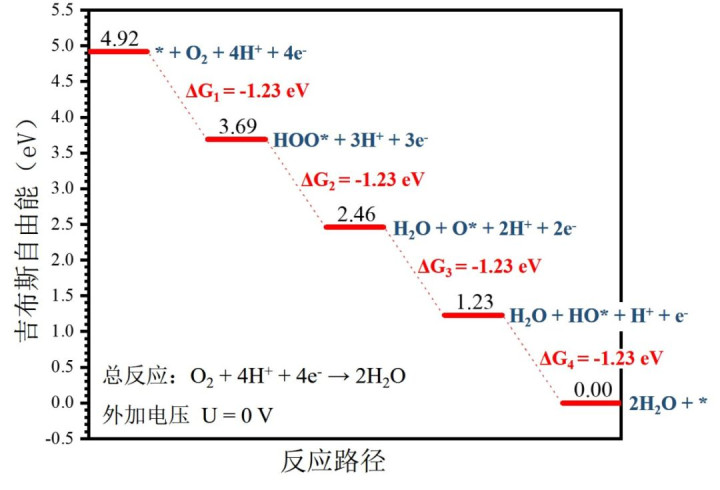
二、DFT计算ORR的核心参数
1.吸附能计算
·O₂吸附能(E_ads):反映催化剂对O₂的吸附能力,过强导致脱附困难,过弱则无法有效吸附。
·中间体吸附能:O*、OOH*、OH*的吸附能与催化剂表面电子结构密切相关。
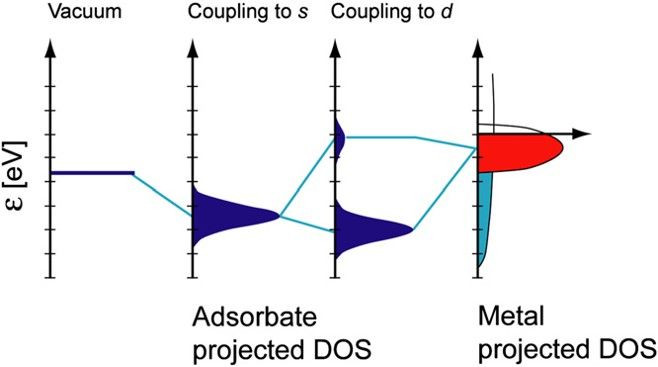
2.d带中心理论
·d带中心(ε_d):过渡金属d轨道能级的平均位置,低于费米能级的ε_d越高,其对小分子吸附越强。
·轨道杂化劈裂:金属与小分子轨道杂化劈裂成成键与反键轨道,反键轨道越高其被电子占据越少则键级越高吸附越强。
3.过电位(η)
·定义:实际反应电位与理论平衡电位的差值,η越小,催化活性越高。
·计算:U=0V时,通过各步骤ΔG的最小值(ΔG_min)得到:η = (1.23-ΔG_mineV) /e(酸性条件)。
三、DFT计算流程与关键设置
1.模型构建
·表面模型:选择金属(如Pt)或合金表面(如Pt₃Co),进行几何结构优化。
·吸附位点:测试不同位点(顶位、桥位、空位)对氧的吸附构型。
2.计算参数
·泛函选择:推荐GGA-PBE,根据需要考虑溶剂化效应(隐式溶剂模型如VASPsol)。
·k点密度:表面模型根据a、b实际长度建议≥2×2×1。
·自旋极化:涉及过渡金属或带氧吸附中间体需开启自旋极化计算。
3.自由能校正
·振动熵校正:通过频率计算获取中间体的振动熵贡献。
·pH与电压校正:采用计算氢电极(CHE)模型,将电位转换为RHE标度。
四、催化剂设计原则
1.合金化策略:
引入Co、Ni等元素调节Pt的d带中心,优化O*吸附能(如Pt₃Co活性优于纯Pt)。
2.单原子催化剂(SACs):
孤立金属原子(如Fe-N-C)通过配位环境调控中间体吸附强度。
3.应力与应变效应:
压缩应变降低ε_d,减弱O*吸附(如核壳结构Au@Pt)。
五、常见误区与解决方案
1.忽略溶剂化效应:
使用隐式溶剂模型或显式水分子层修正吸附能计算模型。
2.未考虑自旋极化:
几何优化需考虑开启自旋极化,避免能量误差。
3.U值校正缺失:
用PBE泛函计算时,对氧化物催化剂(如MnO₂),需加Hubbard U修正强关联效应。
结语:DFT计算为ORR催化剂设计提供了原子尺度的洞察力,但需紧密结合实验验证。未来,机器学习加速的“计算-实验闭环”将推动高效催化剂的发现!